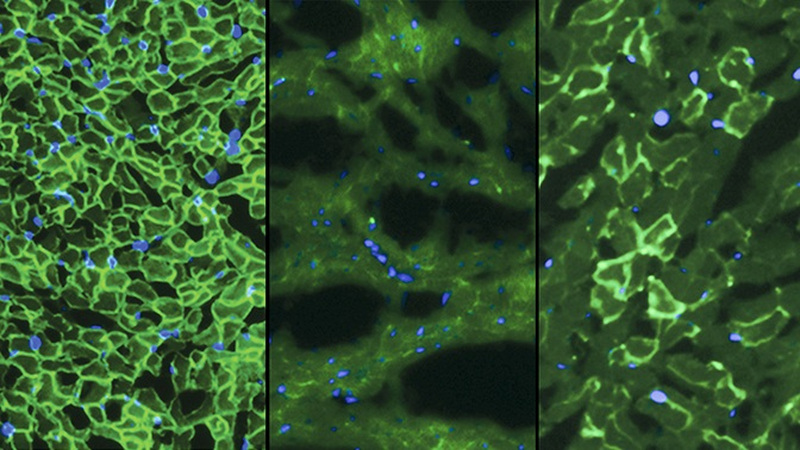
Per la prima volta, gli scienziati hanno utilizzato il CRISPR gene-strumento di editing per trattare con successo una genetica disturbo muscolare, in un mammifero adulto. Si tratta di un promettente innovazione medica che potrebbe presto portare a umana terapie.
Gli scienziati hanno lottato per il trattamento della distrofia muscolare di Duchenne, per decenni. Ad oggi, i loro sforzi si sono concentrati sul trattamento di cellule coltivate in piastre di petri, o nel tentativo di ottenere CRISPR/cas9, un potente DNA di taglia-e-incolla strumento, per fornire riparato copia dei geni difettosi, in modo efficace e sicuro. Ora, con un appositamente modificata del virus, i ricercatori della Duke University hanno confermato una soluzione promettente usando i modelli di mouse. Il team di studio compare nell’ultimo numero di Science.
La distrofia muscolare di Duchenne è una malattia di deperimento muscolare che colpisce uno in 5.000 neonati maschi. La genetica glitch è sul cromosoma X, quindi ragazze con due cromosomi X tendono ad avere almeno una copia funzionante del gene. I sintomi compaiono tipicamente di età compresa tra i 3 e 5, dopo di che la malattia progredisce rapidamente. La maggior parte dei ragazzi sono in grado di camminare per il momento sono 12, e alla fine hanno bisogno di un respiratore per respirare. L’aspettativa di vita è tra i 20 ai 30 anni.
La malattia genetica è causata dall’assenza di distrofina, una fondamentale lunga catena proteica che mantiene l’integrità dei muscoli. La distrofina è codificata da un gene che contiene circa l ‘ 80 proteina-codifica regioni, chiamati esoni. Se anche un singolo esone è male mutato, la catena non costruita. E senza distrofina, i muscoli che lentamente si deteriora.
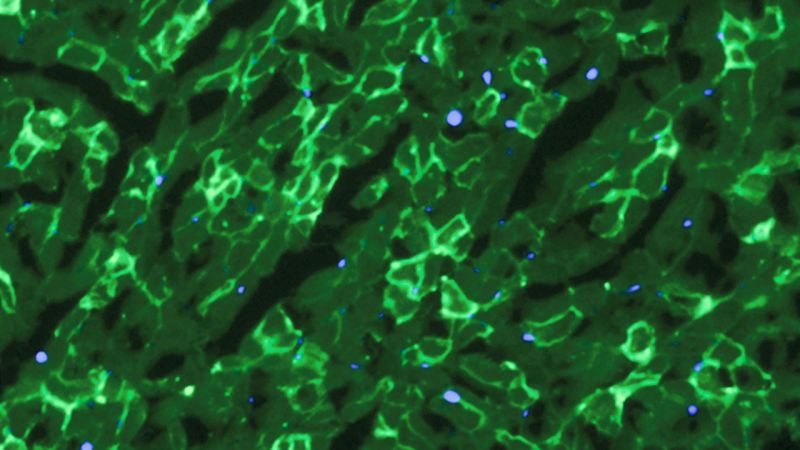
Restaurato produzione di distrofina (luce verde) nelle cellule muscolari di topi. Credito: C. E. Nelson et al., 2015
Il Duca di ricercatori, guidato dal genetista Chris Nelson, utilizzato CRISPR/cas9 per rimuovere la problematica del DNA che è stato impedisce alle cellule di produrre distrofina.
Sponsorizzato
CRISPR, uno strumento che è emerso solo tre anni fa, permette agli scienziati di modifica genomi con incredibile precisione e flessibilità. Come una persona che cerca di risolvere un puzzle, il sistema utilizza un DNA sintetico noto come CRISPRs per la scansione del genoma alla ricerca del posto giusto. Una proteina chiamata cas9 agisce come un paio di forbici per tagliare il DNA.
Per fornire queste alterazioni genetiche, il Duca, i ricercatori hanno utilizzato un tipo di non-virus patogeni. “Un grande ostacolo per la gene editing è la consegna. Sappiamo che i geni hanno bisogno di essere corretti per alcune malattie, ma di ottenere il gene editing strumenti di cui hanno bisogno per andare è una sfida enorme”, ha detto Nelson in un comunicato. “Il modo migliore che abbiamo a che fare adesso è quello di approfittare di virus, perché hanno speso miliardi di anni di evoluzione per capire come ottenere i loro propri geni virali nelle cellule.”
Per lo studio, i ricercatori hanno lavorato con topi geneticamente modificati che aveva debilitante mutazione in uno degli esoni del gene della distrofina. Gli scienziati hanno programmato il nuovo CRISPR/cas9 sistema per eliminare le disfunzioni esone, lasciando naturale del corpo di riparare il sistema per il punto rimanente gene di nuovo insieme. Il risultato è stato accorciato, ma funzionale, la versione del gene.
Al fine di raggiungere ogni muscolo, il virus è stato iniettato nel sangue dei topi. I risultati hanno mostrato misurabili correzioni di muscoli in tutto il corpo, compreso il cuore—di un risultato particolarmente importante, considerando che l’insufficienza cardiaca è la principale causa di morte tra i pazienti Duchenne.
Tuttavia, i topi che hanno ricevuto la terapia non fare come topi normali su prove muscolari, quindi non è una cura. Detto questo, i ricercatori ritengono che c’è un sacco di spazio per migliorare, e che oltre l ‘ 80 per cento delle persone con DMD potrebbe beneficiare di avere un guasto esone rimosso.
“C’è ancora una notevole quantità di lavoro da fare per tradurre questo per una terapia umana, e dimostrare di sicurezza,” ha detto il Duca ricercatore Charles A. Gersbach. “Ma questi risultati provenienti dai nostri primi esperimenti sono molto interessanti. Da qui, ci sarà l’ottimizzazione del sistema di distribuzione, valutando la questione più grave modelli di DMD, e di valutare l’efficienza e la sicurezza in animali di grandi dimensioni con l’obiettivo finale di ottenere in studi clinici.”
Altre due squadre—tutti a lavorare in modo indipendente l’uno dall’altro, ha raggiunto risultati simili nella loro ricerca. Questi studi sono stati condotti da Eric Olson presso l’Università del Texas Southwestern Medical Center e Amy Scommessa all’Università di Harvard.
A differenza di sforzi per modificare la linea germinale di embrioni, questo particolare approccio può essere applicato a un essere vivente. Questo significa che i cambiamenti genetici possono essere introdotti più tardi nella vita, e non sono ereditarie.
“La recente discussione sull’utilizzo CRISPR per correggere le mutazioni genetiche negli embrioni umani ha giustamente suscitato notevole preoccupazione per le implicazioni etiche di tale approccio,” ha detto Gersbach“, Ma di utilizzare CRISPR per correggere le mutazioni genetiche nei tessuti colpiti di malati, non è in discussione. Questi studi mostrano un percorso in cui ciò è possibile, ma c’è ancora una notevole quantità di lavoro da fare.”
[Duke University, New York Times, la Scienza AAAS]
E-mail all’autore george@gizmodo.com e a seguirlo a @dvorsky. Immagine in alto: Distrofina (luce verde) è visto nel muscolo cardiaco di topi normali (a sinistra), manca in topi con DMD (al centro), e parzialmente ristrutturato nel Duchenne topi trattati con CRISPR/Cas9 (a destra). Credito: C. E. Nelson et al., 2015